Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
C
Canonical molecular
orbital (synonymous with self-consistent
field orbitals) - The molecular
orbitals which produce a Fock matrix in the canonical (diagonal)
form. These orbitals are delocalized over the whole molecule and
form the basis for an irreducible representation of the point
group defined by the symmetry of the molecule.
Characteristic polynomial
- is defined for an arbitrary square matrix A of order
n as
Pn(x) = det(xEn
- A)
where En is the unit matrix of order n
and det stands for the determinant of the matrix. The chemical term
equivalent to characteristic polynomial is a secular polynomial.
Charge decomposition analysis
(CDA) - A fragment molecular
orbital partioning scheme for analyzing the electronic interactions
between closed-shell fragments A and B in terms of donation A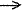 B, backdonation A
B, backdonation A  B, and repulsive polarization A
B, and repulsive polarization A  B. The three terms are given by the mixing of the occupied orbitals
of the fragments (donation and backdonation), and by mixing of the occupied
orbitals of both fragments (repulsive polarization). The rest term D,
which is given by the mixing of the unoccupied orbitals of the fragments,
is a probe which shows if A-B may be considered a donor-acceptor bond.
DAPPRICH and FRENKING (1995).
B. The three terms are given by the mixing of the occupied orbitals
of the fragments (donation and backdonation), and by mixing of the occupied
orbitals of both fragments (repulsive polarization). The rest term D,
which is given by the mixing of the unoccupied orbitals of the fragments,
is a probe which shows if A-B may be considered a donor-acceptor bond.
DAPPRICH and FRENKING (1995).
Charge density - see Electron
density.
Charge-transfer complex
- A ground state adduct
which exhibits an observable charge transfer absorption band. IUPAC
PHYSICAL ORGANIC CHEMISTRY GLOSSARY (1994).
Chemical bond - When forces acting
between two atoms or groups of atoms lead to the formation of a stable
independent molecular entity, a chemical bond is considered to exist
between these atoms or groups. The principal characteristic of a
bond in a molecule is the existence of a region between the nuclei of
constant potential contours that allows the potential energy to improve
substantially by atomic contraction at the expense of only a small increase
in kinetic energy. Not only directed covalent
bonds characteristic of organic compounds, but also bonds such
as those existing between sodium cations and chloride anions in a crystal
of sodium chloride or the bonds binding aluminium to six molecules of
water in its environment, and even weak bonds that link two molecules
of O2 into O4, are to be attributed to chemical
bonds. PAULING (1960); RUEDENBERG
(1962), SUTCLIFFE (1992).
See also Coordination, Covalent
bond, Dative bond, Hydrogen
bond, Ionic bond, Van
der Waals systems.
Chirality - The property of an
object due to which it cannot be superimposed on its mirror image by
translation or rotation. A molecule is chiral if it lacks Sn
mirror-rotational axes. The notion of chirality has also been extended
to displacements in space and other processes. A chiral process consists
of successive states all of which are chiral.
Chromatic graph - see Molecular
graph.
Closed shell molecular systems - Even-electron
atomic or molecular systems whose
electron configurations consist
of doubly occupied orbitals.
Complete Active Space (CAS) -
A computational scheme employed in Multiconfiguration
SCF methods
especially suitable for studies of excited
states. The wavefunction
is defined by selecting a set of active orbitals,
and is constructed as a linear expansion in the set of configuration
functions that can be generated by occupying the active orbitals
in all ways consistent with an overall spin and space symmetry (full
Configuration Interaction).
ROOS (1987)
Complete active space Self-consistent
field second-order perturbation theory (CASPT2) - Theoretical scheme
that in the first step takes electron
correlation into account only to a certain extent by using a
CAS formalism, while the remaining electron correlation is included
through the use of of second-order
perturbation theory.
Complete basis set (CBS) theory
- Set of methods developed for computing very accurate energies. These
methods use extrapolations of the Hartree-Fock
and the second order correlation energies
to the complete basis set limit.
Computational chemistry
- Those aspects of molecular research that are expedited or rendered
practical by computers. LIPKOWITZ and BOYD
(1990).
Concerted reaction - A
single step reaction through which reactants are directly transformed
into products, i.e. without involvement of any intermediates. WOODWARD
and HOFFMANN (1969).
Configuration interaction
(CI) - The mixing of many-electron wavefunctions
constructed from different electronic configurations to obtain an improved
many-electron state. In the CI method, an n-electron wavefunction
is expanded as a linear combination of Slater
determinants. CI proceeds by constructing other determinants
through replacing one or more occupied orbitals
within the Hartree-Fock determinant (see Hartree-Fock
method) with a virtual orbital. In the full CI method, the
wavefunction Y is represented by a linear
combination of the Hartree- Fock determinant Y0
and all possible substitutions Yi
. The ci are the set of coefficients to be solved
for by minimizing the energy of the resultant wavefunction.
In practical calculations, CI methods are usually used
which augment the HF method by only a limited set of substitutions:
the CIS method adds single excitations to the HF determinant, CID adds
double excitations, CISD adds single and double excitations, and
so on. FORESMAN and FRISCH (1996); SCHAEFER
(1972).
Conical intersection (synonymous
with the term "funnel") - An n-2 dimensional subspace of n
nuclear coordinates in which two electronic
states are degenerate. Such type potential
energy surface crossings of two singlet (or two triplet)
surfaces or singlet-triplet surface intersections provide a very
efficient channel for radiationless
transition or for chemical transformation occurring from
the lowest excited state of
polyatomic molecular systems, even in the case when two electronic
states possess the same symmetry (contrary to the noncrossing
rule, which is rigorously valid only for diatomics). BERNARDI,
OLIVUCCI, and ROBB (1996); TELLER
(1969).
Conjugation - In a topological
sense, the indication that each pair of multiple (double or triple)
bonds in a polyunsaturated molecule is separated by one single (or
double) bond. In a conjugated system one multiple bond can be replaced
by a centre with a fully or incompletely occupied orbital, (i. e. an
atom with a lone pair, an unpaired electron, or a vacant orbital).
The term conjugation was extended to orbital language to describe
particular orbital interactions
(p-conjugation, s-conjugation)
given by the topology of the molecule. Conjugation implies an alternation
between stronger and weaker orbital interactions leading to a corresponding
alternation of the resonance
integrals. CREMER (1988).
Conservation of orbital symmetry
- The orbital symmetry control of concerted
reactions; this requires transformation of the molecular
orbitals of reactants into those of products to proceed
continuously by following a reaction
path along which the symmetry of these orbitals remains
unchanged. Reactions which adhere to this requirement are classified
as symmetry allowed reactions,
and those which do not as symmetry
forbidden reactions. WOODWARD and
HOFFMANN (1969).
Consistent Force Field (CFF)
- A force field for calculating
structures, energies and vibration frequencies of both small organic
molecules and large biomolecular systems, including peptides, proteins,
nucleic acids, carbohydrates and lipids. The CFF differs from empirical
force fields in that its force constants are derived from a rigorous
quantum mechanical procedure. MAPLE
(1998).
Coordination - The bonding of a Lewis
base to a Lewis acid via a dative
or coordinate covalent bond.
Coordination number of an atom
- The number of atoms by which that atom is directly surrounded,
and to each of which it is attached by the direct sharing of electrons.
Core approximation (synonymous
with Pseudopotential
approximation) - The approximation treating
atoms as being subdivided into a core composed of a nucleus and tightly
bound inner electrons (which form a cloud of negative charge representing
the time averaged motion of inner electrons) and a valence shell
of electrons that are less tightly bound and may be delocalized over
a molecular entity. This simplification allows one to greatly reduce
the number of electrons that must be taken into account in the calculation
of molecular properties. It is used in all semiempirical
quantum mechanical methods and in some of ab
initio quantum mechanical methods.
Correlation consistent basis
sets - Sets of contracted gaussian functions specially designed
to account as much as possible for the correlation
energy of valence electrons. DUNNING,
PETERSON, and WOON (1998)
Correlation diagram - A diagram
which shows the relative energies of orbitals, configurations, valence
bond structures or states of reactants and products of a reaction as
a function of molecular geometry or another suitable parameter.
Correlation energy - The difference
between the energy of a system calculated as the minimal value within
the Hartree-Fock approximation (see Hartree-Fock
limit) and the exact nonrelativitistic energy of that system.
The correlation energy arises because the Hamiltonian
in the Hartree-Fock method
includes an averaged interelectronic potential which does not account
for the electron correlation
in a molecular system.
Coulomb integral (see Coulomb
repulsion). In the Hückel
molecular orbital theory , is treated as an empirical parameter
supposed to have a value am
characteristic of the AO fm
and the atom of which it is an AO, and independent of the rest of
molecule. In the HMO theory, it is usually assumed that am
is equal to ionization potential
of an electron occupying the AO fm
of the corresponding atom. DEWAR (1969)
Coulomb repulsion - The potential
energy component corresponding to the electrostatic interaction between
each pair of charged particles:

where eo is the permittivity of
a vacuum, Drij is
the distance between the two particles, and ei and
ej are the charges on particles i and j. In molecular
orbital theory, the electrostatic repulsion between the
two electrons occupying the orbitals yi
and yj. In the Hartree-Fock
method the mean Coulomb repulsion is determined by the value
of the Coulomb integral

See also Exchange repulsion.
Counterpoise correction
- Approximate method to estimate the value of the basis
set superposition error.
For a complex A-B formed by two interacting monomers, A and B, the BSSE
is estimated as the difference between monomer energies with the regular
basis set of each monomer and the energies calculated with
the full basis functions for the whole complex. The latter energies
for each monomer are calculated by adding to the normal basis set the
basis functions of the other monomer located at the corresponding nuclear
positions, but without the nuclei present. These additional basis are
called ghost orbitals.
Coupled cluster (CC) method -
An ab initio quantum mechanical
method in which electron
correlation effects are introduced by acting with the exponential
operator exp(T) on the zero-order
wavefunction,
and where T consists of the sum of all possible excitation operators.
A CCSD(T) calculation provides for a higher-level treatment of electron
correlation beyond MP4 (see M�
ller-Plesset Perturbation Theory). FORESMAN
and FRISCH (1996).
Covalent bond - A type of
bonding associated with the sharing of two electrons usually between
two atomic centers of a molecular entity. The mechanism of covalent
bonding that causes decrease in the total potential energy of a
molecule as its constituent atoms move towards each other is orbital
contraction, which increases the localization of the electrons around
the nuclei and decreases the accumulated charge in the bonding region(s).
BACSKAY, REIMERS, and NORDHOLM (1997);
RUEDENBERG (1962).
Crystal field - The average static
electric field experienced by an ion, molecule or atom in a crystal
generated by all the other surrounding atoms, molecules, or ions.
The concept introduced by Becquerel and developed by Bethe has been
extended to refer to the field generated by the ligands (anions
or neutral species) surrounding a transition metal ion in a coordination
compound. With the understatement that some covalency (see Covalent
bond) may exist in the metal-ligand bond, it is often referred
to as ligand field.
Crystal orbital (synonymous with
band orbital) - One-electron
function extended throughout a crystal. Typically a linear combination
of Bloch orbitals:

The Bloch orbitals incorporate the translational symmetry by application
of boundary conditions:

where cm(r) is the n-th atomic
orbital at the unit cell defined
by the translation vector r. The different values adopted by
the wave vector k determine
the symmetry and nodal properties of the crystal orbital.
Crystal orbital overlap population
(COOP) - The overlap population weighted density
of states. For a solid, COOP serves as the
differential version of the bond
order concept. The integral of the COOP curve up to the
Fermi level
is the total overlap population of the specified bond in
a solid, whereas the value of COOP(E)dE represents the
contribution to the total overlap population of those crystal
orbitals whose energy levels lie in the range of E and E+dE.
HOFFMANN (1988).
Curve-crossing model - Model
of organic reactivity that generates a reaction
profile from curves that describe the energies of reactant,
product, and intermediate electronic
configurations (or, alternatively, reactant, product and
intermediate electronic states)
as a function of the reaction
coordinate. The crossing
reflects the electronic reorganization that accompanies the transformation
of reactants and products. PROSS (1995);
PROSS and SHAIK (1983), SHAIK
(1981).
