Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
S
Saddle point - A point of
lowest maximum energy on a valley (reaction
path) connecting two minima on the potential
energy surface. In mathematical terms, the stationary point
on the PES in which the Hessian
matrix has only one negative eigenvalue is assigned to the saddle
point of the first rank. The saddle point is associated with the
transition state
structure.
Scaling factor - A variational
parameter (see variational
method) used as a multiplier of each nuclear Cartesian and
electronic coordinate chosen to minimize the variational integral and
to make a trial variation function to satisfy the virial
theorem. In practical calculations, the numeral factor to
scale computed values, e.g. vibrational frequencies, to those found
in experiments.
Second-order Jahn-Teller
(SOJT) effect - see Jahn-Teller
effect.
Secular equation - In molecular
orbital theory, the determinant of matrix elements whose solution
leads to the set of energy levels of molecular
orbitals for a given system.
Selection rule - A rule which states
whether a given transition or process is allowed or forbidden (i.e.
its intensity is high or zero, or close to zero) on the basis of
the symmetry or spin of the wavefunctions
of the initial and final states.
Self-Consistent Field (SCF)
method - see Hartree-Fock
method.
Self-Consistent Reaction
Field (SCRF) methods - Methods designed to treat solvation effects
which incorporate the essential elements of the reaction field theory
in the conventional SCF approach. The classical reaction field is treated
as a perturbation, so that the hamiltonian is given by
HSCRF = H0 + HRF
Semiempirical quantum mechanical
methods - Methods which use parameters derived from experimental
data to simplify computations. The simplification may occur at various
levels: simplification of the Hamiltonian
(e.g. as in the Extended Hückel
method), approximate evaluation of certain molecular integrals
(see, for example, Zero differential
overlap approximation), simplification of the wavefunction
(for example, use of a Pi
(p) electron approximation
as in Pariser-Parr-Pople
method) etc. MURRELL and HARGET
(1972); POPLE and BEVERIDGE (1970).
Singly occupied molecular orbital
(SOMO) - see Frontier
orbitals.
Size consistency - A property
which guarantees that the method scales properly with the number of
particles, i.e., when doubling the size of the system investigated and
keeping the particle density constant, the energy of the system should
double.
Slater determinant - The
determinantal representation of many-electron wavefunction
Y which conforms to the requirement of
the antisymmetry principle.
Slater type atomic orbital (STO) -
The exponential function centered on an atom; its radial dependence
is given by Nrn-1exp(-zr),
where n is the effective principal quantum number and z
is the orbital exponent (screening constant) derived from empirical
considerations. The angular dependence is usually introduced by
multiplying the radial one by a spherical harmonic glm(qF).
Soliton - Vibration coupled with longitudinal
sound waves propagated as localized quasiparticles. From a chemical
point of view, a soliton can be regarded as a migrating region of conformational
change.
Solvation energy - The change
in Gibbs energy when an ion or molecule is transferred from a vacuum
(or the gas phase) to a solvent. The main contributions to the solvation
energy come from: (a) the cavitation energy of formation of the
hole which preserves the dissolved species in the solvent; (b) the
orientation energy of partial orientation of the dipoles; (c) the isotropic
interaction energy of electrostatic and dispersion origin; and (d)
the anisotropic energy of specific interactions, e.g. hydrogen
bonds, donor-acceptor interactions etc.
Spin conservation rule - see
Wigner rule.
Spin contamination - In unrestricted
Hartree-Fock method,
the wavefunctions obtained
are eigenfunctions of the Hamiltonian
and the spin projection Sz
operators, but not eigenfunctions of the S2
operator. As a result, the wavefunctions of the doublet systems are
spin contaminated to some extent by admixtures of quartet, sextet,
and higher spin states. The eigenvalues of the S2
operator are given as a measure of the spin contamination. LÖWDIN
(1966).
Spin crossover - A type of molecular
magnetism that is the result of electronic instability (see electronic
stability) caused by external constraints (temperature, pressure,
or electromagnetic radiation), which induce structural changes
at molecular and lattice levels. The phenomenon is most characteristic
of first-row transition metal complexes, e. g. those of Fe(II). An example
of spin- crossover complexes (the term of spin-state
isomers is also used) is [Fe(2-pic)3]Cl2
. EtOH (2-pic = 2-picolylamine). At the Fe-N distance
of 203.2 pm (115K), the complex has an electronic low-spin
state (1A1), whereas stretching the bond
up to 219.9 pm at 227 K induces the transition to a high-spin
state (5T2). GÜTLICH,
GOODWIN, and HENDRICKSON (1994).
Spin density - The excess
of the electron density
related to the electron with a spin over
that of the electron with b spin (see
spin-orbital) at a given point of
an open-shell system.
For a closed-shell system
spin density is zero everywhere.
Spin polarization - Static
and dynamic spin polarization
effects are distinguished. The static polarization of an electron
spin occurs in the C-H bonds of aromatic radicals where the
s-electron closest to an unpaired p-electron
tends to have its spin parallel to that of the p-electron.
Likewise, static spin polarization arises in the p-system
of conjugated radicals: the electron of the doubly occupied molecular
orbital prefers to take a closer spatial position to the odd one, which
spin is parallel to the latter. The effect reflects the energy unfavourable
situation if an electron of opposite spin were to come nearby. The
dynamic spin polarization is the instantaneous electron
correlation effect occurring in biradicals
(e.g. in 90o-twisted ethene, cyclobuta-1,3-diene) leading
to correlation of spins of electrons in inner-shell orbitals with those
of odd electrons. Essentially, a molecule seeks out a local (p,s)
or (p,p) triplet configuration at each center
(as is the case of static spin polarization)
to decrease the overall coulomb
repulsion energy of singlet states. The effect is termed dynamic
since spin-polarized electron
configuration has a counterpart where all the spins are reversed.
BORDEN (1982).
Spin projection - A component,
MS , of the angular spin moment S along
an arbitrary axis (usually chosen as the z-direction). MS
can take the values between -S and S: -S, -S+1,
..., S-1, S. The term is also used to denote an operation
(spin-symmetry projection) allowing one to eliminate spin
contamination in the cases where wavefunctions
are not eigenfunctions of the operator S2.
Spin-coupled (SC) wavefunction
- Representation of a wavefunction
in the modified valence bond
theory. A spin-coupled wavefunction
describes a molecular system with a total number of electrons Nt
by subdividing these into inactive core electrons placed in doubly occupied
orbitals and N active
(valence) electrons placed in N distinct, singly occupied, nonorthogonal
orbitals, the spins of which are coupled together in all allowable
ways to form the required overall resultant S. GERRATT,
COOPER, KARADAKOV, and RAIMOND (1997).
Spin-orbit coupling - The
interaction of the electron spin magnetic moment with the magnetic moment
due to the orbital motion of the electron, and the consequent mixing
of electronic states of
different multiplicity.
Spin-orbital - The complete one-electron
wavefunction given (in the
absence or in the case of neglect of spin-orbit
coupling) by a product of a spatial function (orbital)
and a spin function. An orbital yi(r)
may be associated with either a(x)
or b(x) spin functions,
the spin coordinate x taking on one of
two possible values (1/2 or -1/2) that measure the spin angular momentum
component along the z-axis in the h/2p
units. This gives rise to the spin-orbitals yi(r)a(x)
and yi(r)b(x)
.
Spin-spin coupling - a small
relativistic effect due to
interaction between the spin magnetic moments of electrons or nuclei.
Spin-state isomers -
see Spin crossover.
Split valence basis set -
see Basis set.
Static spin polarization - see
Spin polarization.
Stationary point (synonymous
with critical point) - On the potential
energy surface
a point at which the energy
gradients with respect to all coordinates vanish.
Stationary state -
A state in which the expectation values of properties do not change
with time. It is given by a wavefunction
representing one of the solutions of the time-independent Schroedinger
equation.
Stereochemical nonrigidity
- The capability of a molecule to undergo fast and reversible intramolecular
isomerization, the energy barrier to which is lower than that allowing
for the preparative isolation of the individual isomers at room
temperature. It is conventional to assign to the stereochemically
nonrigid systems those compounds whose molecules rearrange rapidly enough
to influence NMR line shapes at temperatures within the practical
range (from –100oC to 200oC) of experimentation.
The energy barriers to thus defined rearrangements fall into the range
of 5-20 kcal/mol (21-85 kJ/mol). A more general term for this phenomenon
is structural nonrigidity.
MUETTERTIES (1970); HOLM
(1975).
See also Automerization,
Fluxional molecules,
Pseudorotation.
Steric energy - see Strain
energy.
Strain energy -The excess energy
due to steric strain of a molecular entity or transition
state structure, i.e. distortions relative to a reference
(real or hypothetical) "strainless" structure with the standard bond
lengths, bond angles and dihedral angles. The strain energy
components involve the following destabilizing terms: non-bonded
repulsions, bond-angle distortions, bond stretch or compression,
rotation around or twisting of double bonds, and electrostatic strain.
In general, the contributions of these components are inseparable
and interdependent. A quantitative assessment of strain and strain
energies can be made by taking the difference between the heat of formation
of the substance under consideration and that of a hypothetical
strain-free model. Several approaches to the assessment of strain
energies have been developed based on the use of energies of isodesmic
and homodesmotic reactions
and on the so-called "strainless increments", i.e. heats
of formation of certain groups (CH3, CH2, CH, C etc). A
synonymous term is steric energy.
GREENBERG and LIEBMAN (1978).
Structural nonrigidity
- see Stereochemical nonrigidity.
Structural stability - Within
the Born-Oppenheimer
(adiabatic) approximation, this is associated with an energy
minimum on a potential energy surface.
Structural stability implies that any change in the coordinates
of the nuclei can only increase the total energy.
Sudden polarization - The
occurrence of a large intramolecular charge separation in the singlet
excited state of polyenes
and their derivatives twisted about a double bond. Unsymmetrical substitution
or geometrical distortion is effective in polarizing the system. An
example is the stabilization of the zwitterionic structure of 90o
twisted ethene (ethan-2-ylium-1-ide) with one methylene group pyramidalized:
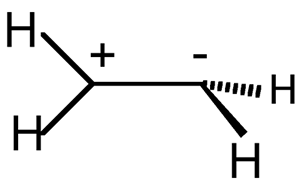
SALEM (1971).
Supermolecule - A discrete oligomolecular
species that results from the intermolecular association of its components.
Supramolecular chemistry
- A field of chemistry related to species of greater complexity than
molecules, that are held together and organized by means of intermolecular
interactions. The objects of supramolecular chemistry are supermolecules
and other polymolecular entities that result from the spontaneous
association of a large number of components into a specific phase
(membranes, vesicles, micelles, solid state structures etc.) LEHN
(1995).
Surface delocalization
- see Delocalization.
Symmetry allowed reaction - see
Conservation of orbital
symmetry.
Symmetry breaking - Instability
of the wavefunction which
appears when it has a lower symmetry than the nuclear framework. It
is found, for instance, in the allyl radical or in the hydrogen peroxide
radical cation. In the former, the nuclear geometry has a C2v
symmetry, but the ROHF wavefunction has only Cs symmetry.
Symmetry element - A geometrical
entity (mirror plane, proper and improper rotation axes, center of inversion)
with respect to which one or more symmetry
operations may be carried out. COTTON
(1964).
Symmetry forbidden reaction -
see Conservation
of orbital symmetry.
Symmetry point group - (synonymous
with Point group) - All
the symmetry operations that leave a particular object (e.g. molecular
entity or function) unchanged constitute together the symmetry point
group for that object.
Symmetry operation - An operation
associated with a symmetry element
that is carried out on some object and which leaves the object unchanged.
Synchronous reaction -
A concerted reaction
in which all the changes in bonding take place in parallel.
