Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
U-V
Uncertainty (Heisenberg) principle
- The statement that two dynamical variables can be simultaneously
well defined only if their quantum mechanical operators commute. If,
for example the momentum of a particle is known with an uncertainty
of Dp, the position of the particle
cannot be determined with the precision better than h/Dp:
Dpx
Dx � h
Unit cell - The cell chosen
to represent best the symmetry of a crystal lattice. The entire lattice
can be derived from it by translations.The translations selected as
the edges of the plane unit cell are denoted as a and b,
and for a space lattice as a, b, c called the crystallographic
axes. The angles between the edges of the three-dimensional unit
cell are a, b and g.
Unitary group - A group which
consists of unitary matrices of fixed order with matrix multiplication
as the group operation.
Unrestricted Hartree-Fock
(UHF) method - see Hartree-Fock
method.
Valence bond configuration mixing (VBCM)
model - A model that provides a framework for conceptualizing
reactivity trends through building up a reaction
profile from its constituent VB configurations. Key configurations
that are invariably employed are those that best describe reactants
YR and products YP.
PROSS (1995), PROSS
and SHAIK (1983), SHAIK, SCHLEGEL, and
WOLFE (1992).
See also Curve-crossing model.
Valence bond (VB) theory - A method
of approximating the total wavefunction
of a molecule as a linear combination of functions related to certain
pairings of electron spins. Each such function is given by a product
of the atomic orbitals of
the separated atoms. As a first approximation the covalent pairing
scheme is employed, in which pairs of electrons between the atoms have
their spins paired as much as possible. To account for electron
correlation effects, mixing in of functions representing
ionic pairing schemes, in which some of the separated atoms are replaced
by positive and negative ions, is employed.The VB and MO methods,
if carried out with the same basis set
of orbitals, are entirely equivalent if all possible ionic structures
are included in the VB and all possible configurations in the MO.
HIBERTY, KLEIN, and TRINAJSTIC (1990);
SHAIK (1989); SLATER
(1931).
Valence state electron affinity
- The electron affinity
of an atom in its particular electron
configuration.
Valence state ionization
potential (VSIP) - The
ionization potential of an atom in its particular electronic
configuration.
Valence-shell-electron-pair repulsion (VSEPR) theory
- A semiquantitative approach to the prediction of the
geometries of compounds of main-group elements. The basic idea of the
theory is that the geometric arrangement of the bonds around a central
atom depends on the number of electron pairs in its valence shell.
A given number of electron pairs adopt that arrangement which keeps
them as far apart as possible, as a consequence of the operation of
the Pauli exclusion principle.
The arrangements of electron pairs are essentially retained independently
of whether they are unshared pairs, or form single, double or triple
bonds. The theory is concerned with the arrangement of predominantly
covalent bonds around a single
central atom. It does not apply to the compounds with ionic
bonds and to molecules with multicenter
bonds. GILLESPIE (1972), GILLESPIE
and ROBINSON (1996).
van der Waals complexes - Molecular
systems in which the individual parts are held together by forces
other than covalent bonds.
These include ionic complexes (where the dominant attractive force
is of electrostatic origin), complexes with hydrogen
bonds, charge-transfer complexes, and true van der Waals
molecules for which the dominant attractive contribution is the
dispersion energy. HOBZA
and ZAHRADNIK (1988).
van der Waals (VDW) interactions
- Noncovalent interactions (weak as compared to
covalent bonds) due to dipole-induced dipole and dispersion
forces (see dispersion energy)
acting at molecules and atoms. In
molecular mechanics models, the energy contributions from
vdW interactions are commonly treated with potential functions of
the distance, r, between each pair of nonbonded atoms, like
the Lennard-Jones potential:
Vvdw = A/r12 - C/r6
Variational method - See Variational
principle.
Variational principle -
The principle according to which for a molecular system an approximate
wavefunction, when substituted
into the Schroedinger equation, will always yield a higher energy than
the actual energy of the system. The more precise the wavefunction that
is chosen, the closer will the calculated energy be to the true energy.
The computational method using this principle to obtain approximations
to correct wavefunctions is called the variational
method. The method is commonly
restricted to the ground state,
but can be extended to others provided they are orthogonalized to
the (true) ground state.
Variational Transition State
- In variational
transition state theory, the optimized dividing surface (hypersurface
in phase state that separates reactants from products). If not all trajectories
passing through the dividing surface and originated at reactants are
directed to products transition
state theory overestimates
the rate. In variational transition state theory one optimizes the location
of the dividing surface to minimize the rate. TRUHLAR
and GARRET (1984), GARRET and TRUHLAR
(1998).
Variational
Transition State Theory (VTST) - see Variational
Transition State .
Vertical electron affinity -
see Electron affinity.
Vertical ionization potential
- see Ionization potential.
Vibronic interactions, theory
of - An approach to the analysis of molecular properties and molecular
transformations which, unlike the Born-Oppenheimer
approximation, assumes that
electronic states depend strongly on nuclear coordinates.
If stationary electronic states (ground,
first excited etc.) are obtained as solutions of
the Schroedinger equation for fixed nuclei, an accounting for vibronic
coupling terms in the Hamiltonian
(interaction of electrons with nuclear displacements) mixes these electronic
states. This mixing is especially strong in the cases of electronic
degeneracy (see Jahn-Teller
effect) and pseudodegeneracy (pseudo
Jahn-Teller effect).
BERSUKER (1984).
Vibronic transition - A transition
which involves a change in both the electronic and vibrational quantum
numbers of a molecular entity. The transition occurs between two electronic
states, but involves a change in both electronic and vibrational
energy. IUPAC PHOTOCHEMICAL GLOSSARY
(1988).
Virial theorem - Interrelates the
kinetic, T, and potential, V, energy of a system in its
stationary states. The
molecular electronic virial theorem is formulated ( J.Slater ) as follows:
2<Tel> = -<V> - 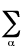
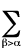 Ra,b(�U/�Ra,b)
Ra,b(�U/�Ra,b)
where Rab is the distance between
nuclei a and b,
U is the potential energy function for nuclear motion and the
sum runs over all internuclear distances. The true wavefunctions
must satisfy the virial theorem. LEVINE
(1970).
Virtual orbital - An orbital from
a set of MOs obtained as solutions of the SCF equations (see Hartree- Fock
method) whose energies are higher than those of doubly occupied
MOs producing the single determinant wavefunction
of lowest energy for a given system. The virtual orbitals obtained from
SCF calculations are not variationally correct approximations (see
variational principle) to
the excited state orbitals.
Their energies are not related to electron
affinities of the molecular system
Volume delocalization -
see Delocalization.
