Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
M
Many body perturbation theory (MBPT)
- In quantum mechanics, methods of perturbation theory
which use an independent particle approximation to obtain the zero-order
wavefunction.
Marcus equation - A relationship
correlating free activation energy, DG#,
with the standard free energy, DGo
of a reaction in the particular solvent
DG# = wr
+ l(1 + DGo’/l)2/4
where DGo’ =
DGo + wp
- wr , wr
(or wp) is the work required to
bring the reactants (or products) together to the mean separation
distance in the transition state
structure, and the vertical reorganization
energy , l, is equal to the fourfold
intrinsic barrier. Originally
developed for treatment of outer-sphere electron transfer reactions,
the Marcus equation has later been found to be applicable to proton
transfer and group-transfer reactions. MARCUS
(1964); SHAIK, SCHLEGEL, and WOLFE (1992).
Maximum hardness, principle of -
A chemical system at a given temperature will evolve to a configuration
of maximum absolute hardness,
h, provided that the potential due to the
nuclei, plus any external potential and the electronic chemical
potential remain constant. In terms of molecular
orbital theory, the highest value of h
reflects the highest possible energy gap between the lowest
unoccupied and highest
occupied molecular orbitals; this value correlates with
the stability (see structural
stability, electronic stability,
thermodynamic stability
and kinetic stability)
of a system. PEARSON (1987); PARR
and CHATTARAJ (1991).
Mesomeric effect (synonymous
with Resonance effect)
- The intramolecular polarization of a conjugated molecular
system brought about by a substituent whose pp
or p-orbitals overlap with the
p-MOs of the conjugated moiety. A distinctive feature of the
mesomeric effect, as compared with the
inductive effect, is that it causes alternation of the
electron density in the conjugated
chain or ring to which the mesomeric substituent is attached. Electron-releasing
substituents with lone electron pairs (e.g. hydroxy, amino groups) are
said to exert positive mesomeric effects, whereas substituents with
low-lying lowest unoccupied molecular
orbitals are characterized by negative mesomeric effects.
Metropolis algorithm - see
Monte Carlo,
method of.
Microscopic reversibility,
principle of - For a system in thermodynamic equilibrium not only
the total number of molecules leaving a given quantum state in unit
time will equal the number arriving in the state in unit time, but
also the number leaving by any one particular path will be equal to
the number arriving by the reverse of that path. The principle was widely
applied to the analyses of reaction mechanisms, in particular of substitution
reactions. In the case of SN2 reactions at tetrahedral centers
implying a formation of the trigonal bipyramid
transition state (or intermediate) structure, the original
formulation of the principle was extended in the following way: if a
molecule or reactant enters a trigonal bipyramid at an apical position,
this (or another) molecule or reactant must likewise leave the trigonal
bipyramid from an apical position. TOLMAN
(1934); WESTHEIMER (1968).
Minimal basis set - see Basis
set.
Minimum energy reaction path (MERP)
- The trajectory orthogonal to the equipotential contours of
a potential energy surface,
which connects the energy minima through a common saddle
point (transition state)
from which it slopes downward along the steepest descent lines in 3N
- 6 configurational space (N is the number of nuclei in the reacting
system). The methodology of MERP allows one to transform the large number
of nuclear coordinates needed to specify the geometry of a polyatomic
system to a unique coordinate. The remaining coordinates vary smoothly
between their values at each end point of the MERP. DEWAR
(1975).
Möbius
system - A cyclic p-conjugated
ribbon-like (see Ribbon
delocalization) delocalized molecular system whose basis
atomic orbitals are organized
in a Möbius strip.
In contrast to Hückel
systems, whose orbital basis sets of a ring have zero or even number
of phase inversions, Möbius
type systems are characterized by an odd number of nodes. The electron
counting rule for the stability of Möbius
systems is opposite to the Hückel
rule: the 4n electron Möbius systems have a closed electron
shell, while the shell of the (4n+2) electron Möbius
systems is open. HEILBRONNER and BOCK
(1976); ZIMMERMAN (1971).
Molecular dynamics (MD), method
of - A method of computational modeling of the static
(equilibrium) and dynamic (kinetic) properties of many-particle systems
by solving numerically the classical Newtonian equation assuming
a predetermined force potential and a protocol for preparation of the
system from a rational starting configuration to monitor the movement
of molecules in liquid or solution, define the phase trajectories
and then average over time, and obtain the magnitudes of the functions
examined. OHMINE and TANAKA (1993);
SIMKIN and SHEIKHET (1995).
See also Monte Carlo, method of.
Molecular electrostatic potential
(MEP) - see Electrostatic
potential.
Molecular entity - A general
term applied to any constitutionally or isotopically distinct atom,
molecule, ion, ion pair, radical, radical ion, complex, conformer etc.,
identifiable as a separately distinguishable entity. IUPAC
PHYSICAL ORGANIC CHEMISTRY GLOSSARY (1994)
Molecular graph - The
graph with differently labeled
(colored) vertices (chromatic graph)
which represent different kinds of atoms and differently labeled
(colored) edges related to different types of bonds. Within the
topological electron distribution
theory, a complete network of the bond
paths for a given nuclear configuration.
Molecular graph theory
- The theory which deals with analyses of all consequences of connectivities
inherent in molecular structure and chemical transformations. The
theory does not produce numerical data but uses available data and
searches for regularities that can be attributed to combinatorial
and topological origins. BALABAN (1976); KING (1983)
See also Molecular graph, Reaction
graph.
Molecular graphics - Rendering
a molecule as output by a computer for display. Sometimes molecular
properties are also shown.
Molecular mechanics (synonymous
with force field method)
- Method of calculation of geometrical and energy characteristics
of molecular entities on the basis of empirical potential functions
(force field) the form of
which is taken from classical mechanics. The method implies transferability
of the potential functions within a network of similar molecules.
An assumption is made on "natural" bond lengths and angles, deviations
from which result in bond and angle strain respectively (see Strain
energy). In the simplest models, the total potential energy,
Vtotal, is broken down into four components:
Vtotal = 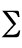 Vr
+ VQ
+
VF +
VvdW
Vr
+ VQ
+
VF +
VvdW
where the sums represent respectively contributions due to bond
stretching and compression terms, valence angle bending terms, internal
rotation or torsional terms and van der Waals interactions. In practice,
electrostatic terms and some other terms (improper torsion or out-of-plane
bending terms, various cross-terms) are also taken into account.
BURKERT and ALLINGER (1982).
Molecular modeling - Generation,
manipulation, and/or representation of molecular structures and associated
physicochemical properties on a computer
Molecular orbital (MO) -
see Orbital.
Molecular orbital (MO) theory
- An approach to molecular quantum mechanics which uses one- electron
functions (orbitals) to approximate
the full wavefunction.
Molecular Rydberg state - An
excited electronic state
which is composed primarily of atomic
orbitals with principal
quantum numbers greater than that of the ground
state and the valence excited states. Such electronic states
typically have a large polarizability.
ROBIN (1974).
MØller-Plesset
(MP) perturbation theory - An approach to
electron correlation which adds higher excitations to Hartree-Fock
method using the technique from many body perturbation
theory. Whereas the first perturbation to the HF energy (MP2
method) will always lower the total
energy, the higher-level MP orders may be positive. They are
also capable of overcorrecting the energy, since the theory is not
variational (see Variational
principle). FORESMAN and FRISCH
(1996).
Monte Carlo (MC), method of - In
mathematics, a method originally used for calculating multiple integrals
by means of a random sample. The method is used for numerical modeling
of many-particle chemical systems, in particular liquids; it is
based on the equilibrium statistical mechanics theory. Observables
A are calculated as mean values over a great number (@
105 - 106) of instant configurations as determined
by coordinates of the particles.
< A>
= 1/N 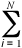 A{ri}
A{ri}
where N is the number of configurations. At the
first stage, various configurations are randomly generated and then
those energetically unrealizable eliminated. An efficient search for
the most probable configurations to be entered into the above
expression is provided by the Metropolis
algorithm based on the principle of Markov’s chain
theory. While being elaborated for the study of equilibrium chemical
systems, MC method is also applied to studies of the dynamics of chemical
reactions.
See also Molecular dynamics,
method of.
Morokuma analysis - An energy
partioning scheme for the interaction energy between atoms or fragments
A and B in the molecule A-B. The interaction energy is given as sum
of three terms: electrostatic (which gives the interaction between the
frozen charge distribution of A and B in the geometry of A-B), exchange
interaction and the orbital relaxation energy (the energy change when
the orthonormal orbitals of the fragments change into the final MO’s
of A-B). The latter term is decomposed further into a polarization term,
which arises from the mixing of occupied and empty orbitals of the same
fragment, and a charge transfer term which gives the mixing of occupied
orbitals of one fragment with the empty orbitals of the other fragment.
The Morocuma analysis is similar to the Extended
transition state method. MOROKUMA
(1977)
Morse potential - The empirical
function relating the potential energy of a molecule to the interatomic
distance r accounting for the anharmonicity (see harmonic
approximation) of bond stretching:
E(r) = De {1 - exp[-a (r
- re)]}2
where De is the bond-dissociation
energy, re is the equilibrium bond
length, and a is a parameter characteristic of a given molecule.
Mulliken population analysis (MPA)
- A partitioning scheme based on the use of density and overlap
matrixes of allocating the electrons of a molecular entity in some fractional
manner among its various parts (atoms, bonds, orbitals).
As with other schemes of partitioning the electron
density in molecules,
MPA is arbitrary and strongly dependent on the particular basis
set employed. However, comparison of population analyses for
a series of molecules is useful for a quantitative description of
intramolecular interactions, chemical reactivity and structural
regularities. MULLIKEN (1955); HEHRE, RADOM,
SCHLEYER, and POPLE (1986).
See also Atomic charge, Bond
order.
Multicenter bond - A type
of chemical bonding for which a localized description requires involvement
of more than two atomic centers. See also Three-center,
two-electron and Three-center,
four-electron bonds.
Multiconfiguration SCF (MC
SCF) method - The configuration
interaction method in which simultaneous optimization of
both the shapes of molecular orbitals
and contributions from different electronic
configurations is carried out by use of the variational
method. The MC SCF method with a large enough set of configurations
allows better estimation of the correlation
energy as compared with the conventional CI method.
Multiplicity - The existence of several
degenerate wavefunctions
distinguished only by the relative orientation of the angular spin
momentum. Defined by the total angular spin momentum S, it is
given by 2S+1.
Multi-reference configuration
interaction (MRCI) - Configuration
interaction method in which the configurations are built by
excitations out of a wavefunction obtained by using multiconfiguration
SCF method.
