Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
D
Dative
bond - The coordination bond formed upon interaction between
molecular species one of which serves as a donor and the other as
an acceptor of the electron pair to be shared in the complex formed,
e.g the N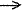 B
bond in H3NBH3.
In spite of the analogy of dative bonds with covalent
bonds in that both types imply sharing a common electron pair
between two vicinal atoms, the former are distinguished by their
significant polarity, lesser strength, and greater length. The distinctive
feature of dative bonds is that their minimum-energy rupture in
the gas phase or in inert solvent follows the heterolytic
bond cleavage path. HAALAND
(1989). see also IUPAC PHYSICAL ORGANIC
CHEMISTRY GLOSSARY (1994).
B
bond in H3NBH3.
In spite of the analogy of dative bonds with covalent
bonds in that both types imply sharing a common electron pair
between two vicinal atoms, the former are distinguished by their
significant polarity, lesser strength, and greater length. The distinctive
feature of dative bonds is that their minimum-energy rupture in
the gas phase or in inert solvent follows the heterolytic
bond cleavage path. HAALAND
(1989). see also IUPAC PHYSICAL ORGANIC
CHEMISTRY GLOSSARY (1994).
Degenerate
electronic states - Electronic
states whose energies are equal.
Degenerate
orbitals - Orbitals whose energy
levels are equal in the absence of external fields. Any linear combination
of the functions corresponding to a given set of degenerate orbitals
serves as an equivalent representation of this set of orbitals.
Degenerate
rearrangement - see Automerization.
Delocalization
- Redistribution of the valence-shell electron density throughout
a molecular entity as compared with some localized models (individual
atoms in their valence states, separated bonds or fragments). Different
topological modes of the electron delocalization include: (a) ribbon
delocalization of either p- or
s-electrons (i.e. electrons occupying respectively
p- and s-orbitals);
(b) surface delocalization
of s-electrons occurring through an overlap
of radially oriented s-orbitals of a cyclic
molecule, as is the case of cyclopropane; and (c) volume
delocalization of
s-electrons through an overlap of s-orbitals
directed inside a molecular polyhedron, as is the case in tricyclo [1.1.0.02,4]
butane (tetrahedrane). DEWAR (1988);
GOLDSTEIN and HOFFMANN (1971).
Delocalization
energy (DE) - The difference between the actual p-electron
energy of a molecular entity and the p-electron
energy of a hypothetical species with a localized form of the p-electron
system. These energies are normally evaluated within Hückel
molecular orbital theory. See Hückel
resonance energy.
Density
functional theory (DFT) - A theory which is concerned with
a quantum mechanical description of atomic and molecular systems
in terms of the electron density.
All properties are functionals of it including the electronic kinetic
energy T[r] and the electron-electron
repulsion energy Vee[r].
The total electronic energy of a given system having N electrons
is expressed as a functional of its single particle density r(r)
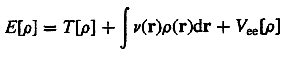
where v(r) is the potential due to the nuclei, is
a minimum when r is the correct ground-state
density. In DFT, the exact exchange term for a single determinant
is replaced by a more general expression, the exchange-correlation
functional, which can include terms accounting for both exchange
repulsion energy and the electron
correlation which is omitted from the Hartree- Fock
method. DFT provides
the conceptual basis to a number of important chemical concepts such
as electronegativity,
absolute hardness and softness,
frontier orbital theory etc.
HOHENBERG and KOHN (1964); PARR
and YANG (1989).
Density
matrix - The one-electron density matrix the elements of
which are defined as
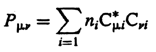
where subscripts m,n are for basis
function, i that of molecular
orbitals, ni is the occupation number of the
ith orbital. The asterisk denotes complex conjugation (required
if the molecular orbitals are not real functions).
Density
of states (DOS) - In a solid, the number of energy
levels in a given energy interval defined as follows:
DOS (E)dE = number of levels berween E and E+dE
In general, DOS (E) is proportional to the inverse of the
slope of the dispersion diagram.The
integral of DOS up to the Fermi
level is the total number of occupied molecular
orbitals in a solid. The DOS curves, therefore, plot the the
distribution of electrons in energy in the solid. HOFFMANN
(1988).
Dewar
resonance energy - see Resonance
energy, various types of.
Diabatic
reaction - Within the adiabatic
approximation, a reaction beginning on one
electron state (ground or excited) potential
energy surface and ending, as a result of radiationless
transition, on another surface.
Diatomics
in Molecules (DIM) - A quantum chemical method based on the assumption
that the energy of molecule may be expressed from diatomic contributions.
TULLY (1980)
See also Atoms in molecules,
theory of.
Differential
overlap, neglect of - see Zero
differential overlap approximation.
Diffuse
functions - Large size versions of s- and p-type atomic
orbitals (as opposed to the normal, contracted functions).
They allow orbitals to occupy a larger region of space; this makes them
important for inclusion into basis
sets for systems where electrons are relatively far from the
nucleus: molecules with lone pairs, anions, systems in their
excited states etc.
See also Basis set.
Dipole
hyperpolarizability - see Hyperpolarizability.
Dipole
moment - The electric dipole of a molecule, m,
is the first derivative of the energy with respect to an applied
electric field. It is a measure of the asymmetry in the molecular charge
distribution and is defined by the relation
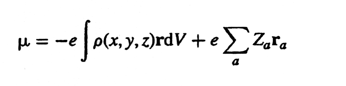
where r is the radius vector of electrons, ra
is the vector from the origin to the nucleus of atomic number Za
and r (x, y, z)
is the electron density. The
dipole moment of a complex molecule can be represented approximately
in the form of the vectorial sum of the moments belonging to the individual
bonds. Dipole moment is independent of origin for a neutral molecule
but origin dependent for an ion. LEVINE
(1970); MINKIN, OSIPOV, and ZHDANOV
(1970)
Dispersion
diagram (synonymous with band diagram)
- Representation of the energy of the
crystal orbitals of a solid as a function of the wave
vector. For three-dimensional solids, such representations
are given for only a few projections of the wave vector, or symmetry
lines of the Brillouin zone.
Dispersion
energy - An attractive component of the energy of intermolecular
interaction resulting from the interaction between the instantaneous,
time-variable dipole of one system and the induced multipole of
the second system. This interaction cannot be interpreted in terms of
classical electrostatics and corresponds to the intersystem component
of the correlation energy.
For two neutral atoms, the dispersion energy is proportional to
the sixth power of the reciprocal distance:
ED 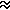 -1/R6.
-1/R6.
HOBZA and ZAHRADNIK (1988)
Distonic
ion - An ion with formally separated charge and radical sites. Distonic
ions may be divided into two distinct classes: those possessing
coordinatively and electronically-saturated charge site (usually
of the onium type) and the ionized biradicals
with two coordinatively-unsaturated sites containing one or three
electrons. YATES, BOUMA, and RADOM (1986).
see also Distonic radical cation (IUPAC
PHYSICAL ORGANIC CHEMISTRY GLOSSARY (1994)).
Double
zeta (DZ basis set) - see Basis
set.
Dynamic
reaction path (DRP) - a classical trajectory method based on molecular
orbital calculations which determines atomic accelerations,
velocities and positions using the energy gradient, and does not
require prior knowledge of the potential
energy surface. STEWART, DAVIS,
and BURGGRAF (1987).
Dynamic
spin polarization (see spin
polarization)
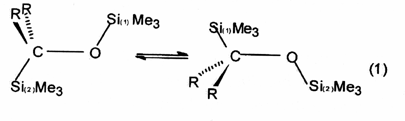
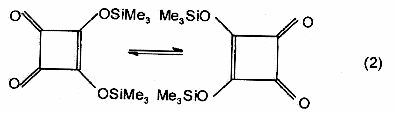
Dyotropic
rearrangement - A class of pericyclic
reactions in which migration of two s-bonded
groups occurs. Two different types of dyotropic rearrangements are
distinguished: (a) those in which two migrating groups interchange
their positions [equation (1)] and (b) those which involve migration
to new bonding sites in a manner that avoids positional exchange [equation
(2)].